




Il premio Lasker Basic Medical Research Award di quest'anno è stato assegnato a Demis Hassabis e John Jumper per il loro contributo alla creazione del sistema di intelligenza artificiale AlphaFold, che prevede la struttura tridimensionale delle proteine in base alla sequenza di primo ordine degli amminoacidi.
I loro risultati risolvono un problema che da tempo tormenta la comunità scientifica e aprono la strada ad accelerare la ricerca in campo biomedico. Le proteine svolgono un ruolo fondamentale nello sviluppo delle malattie: nel morbo di Alzheimer, si ripiegano e si aggregano; nel cancro, la loro funzione regolatrice viene persa; nei disturbi metabolici congeniti, sono disfunzionali; nella fibrosi cistica, si posizionano nello spazio sbagliato della cellula. Questi sono solo alcuni dei numerosi meccanismi che causano le malattie. Modelli dettagliati della struttura proteica possono fornire configurazioni atomiche, guidare la progettazione o la selezione di molecole ad alta affinità e accelerare la scoperta di farmaci.
Le strutture proteiche sono generalmente determinate mediante cristallografia a raggi X, risonanza magnetica nucleare e criomicroscopia elettronica. Questi metodi sono costosi e richiedono molto tempo. Ciò si traduce nell'esistenza di database di strutture proteiche 3D con solo circa 200.000 dati strutturali, mentre la tecnologia di sequenziamento del DNA ha prodotto oltre 8 milioni di sequenze proteiche. Negli anni '60, Anfinsen et al. scoprirono che la sequenza 1D degli amminoacidi può ripiegarsi spontaneamente e ripetutamente in una conformazione tridimensionale funzionale (Figura 1A) e che gli "chaperoni" molecolari possono accelerare e facilitare questo processo. Queste osservazioni aprono la strada a una sfida sessantennale nella biologia molecolare: predire la struttura 3D delle proteine a partire dalla sequenza 1D degli amminoacidi. Con il successo del Progetto Genoma Umano, la nostra capacità di ottenere sequenze amminoacidiche 1D è notevolmente migliorata e questa sfida è diventata ancora più urgente.
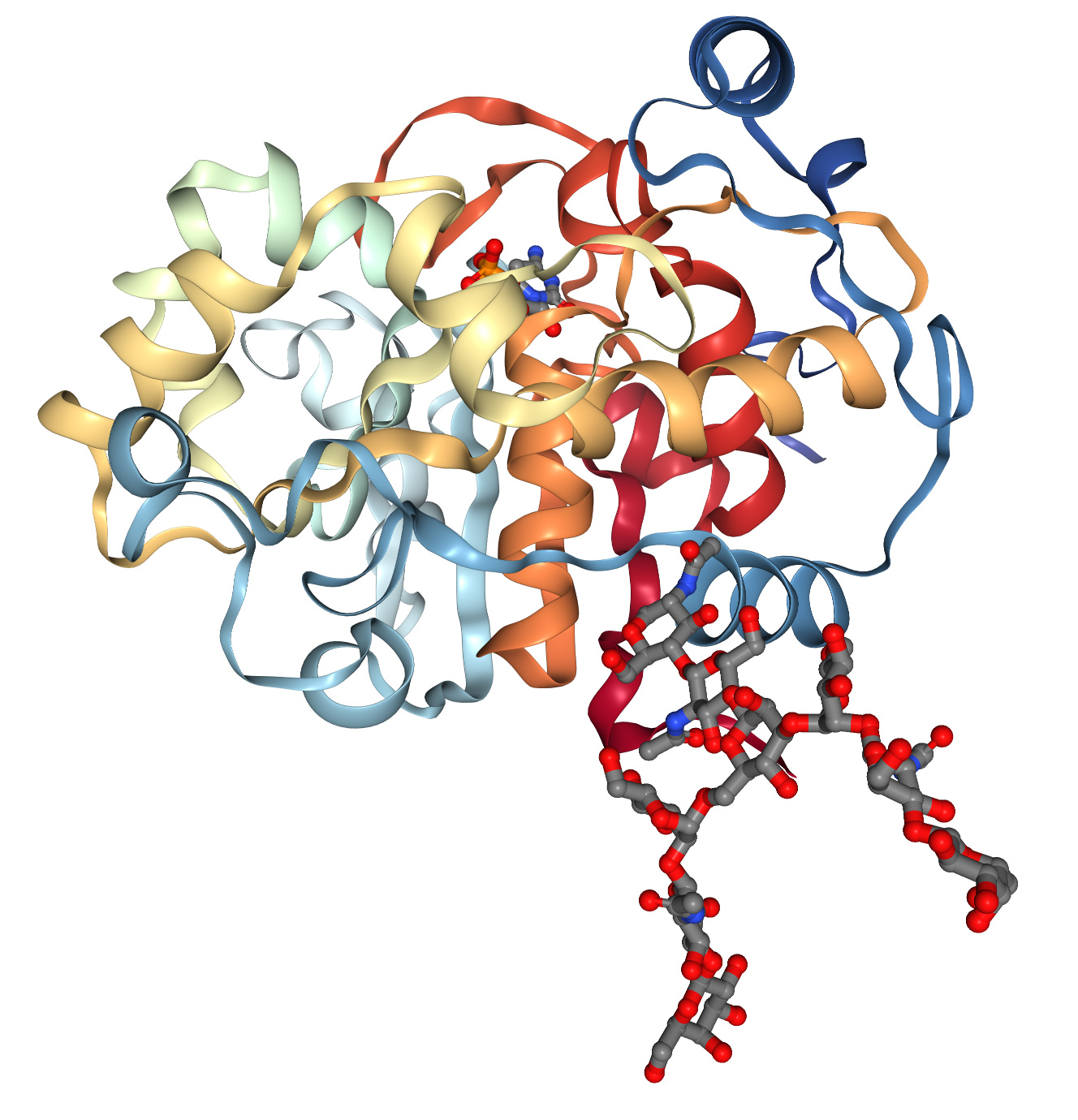
Prevedere le strutture proteiche è difficile per diverse ragioni. In primo luogo, tutte le possibili posizioni tridimensionali di ogni atomo in ogni amminoacido richiedono molta esplorazione. In secondo luogo, le proteine sfruttano al massimo la complementarietà nella loro struttura chimica per configurare in modo efficiente gli atomi. Poiché le proteine hanno tipicamente centinaia di "donatori" di legami idrogeno (solitamente ossigeno) che dovrebbero essere vicini all'"accettore" di legami idrogeno (solitamente azoto legato all'idrogeno), può essere molto difficile trovare conformazioni in cui quasi ogni donatore sia vicino all'accettore. In terzo luogo, gli esempi per l'addestramento di metodi sperimentali sono limitati, quindi è necessario comprendere le potenziali interazioni tridimensionali tra amminoacidi sulla base di sequenze 1D utilizzando informazioni sull'evoluzione delle proteine rilevanti.
La fisica è stata utilizzata per la prima volta per modellare l'interazione degli atomi nella ricerca della conformazione migliore, ed è stato sviluppato un metodo per prevedere la struttura delle proteine. Karplus, Levitt e Warshel hanno ricevuto il Premio Nobel per la Chimica nel 2013 per il loro lavoro sulla simulazione computazionale delle proteine. Tuttavia, i metodi basati sulla fisica sono computazionalmente costosi e richiedono un'elaborazione approssimativa, quindi non è possibile prevedere strutture tridimensionali precise. Un altro approccio "basato sulla conoscenza" consiste nell'utilizzare database di strutture e sequenze note per addestrare modelli tramite intelligenza artificiale e apprendimento automatico (AI-ML). Hassabis e Jumper applicano elementi sia della fisica che dell'AI-ML, ma l'innovazione e il balzo in avanti nelle prestazioni dell'approccio derivano principalmente dall'AI-ML. I due ricercatori hanno combinato in modo creativo grandi database pubblici con risorse di calcolo di livello industriale per creare AlphaFold.
Come facciamo a sapere che hanno "risolto" il puzzle della previsione strutturale? Nel 1994 è stata istituita la competizione Critical Assessment of Structure Prediction (CASP), che si riunisce ogni due anni per monitorare i progressi della previsione strutturale. I ricercatori condivideranno la sequenza 1D della proteina di cui hanno recentemente risolto la struttura, ma i cui risultati non sono ancora stati pubblicati. Il predittore predice la struttura tridimensionale utilizzando questa sequenza 1D, e il valutatore giudica in modo indipendente la qualità dei risultati previsti confrontandoli con la struttura tridimensionale fornita dallo sperimentatore (fornita solo al valutatore). CASP conduce revisioni cieche reali e registra salti periodici di prestazioni associati all'innovazione metodologica. Alla 14a Conferenza CASP del 2020, i risultati di previsione di AlphaFold hanno mostrato un tale balzo in avanti nelle prestazioni che gli organizzatori hanno annunciato che il problema della previsione della struttura 3D era stato risolto: l'accuratezza della maggior parte delle previsioni era prossima a quella delle misurazioni sperimentali.
Il significato più ampio è che il lavoro di Hassabis e Jumper dimostra in modo convincente come l'intelligenza artificiale e il machine learning (IA-ML) possano trasformare la scienza. La ricerca dimostra che l'IA-ML può costruire ipotesi scientifiche complesse a partire da più fonti di dati, che meccanismi di attenzione (simili a quelli di ChatGPT) possono scoprire dipendenze e correlazioni chiave nelle fonti di dati e che l'IA-ML può autovalutare la qualità dei suoi risultati. L'IA-ML è essenzialmente scienza.
Data di pubblicazione: 23 settembre 2023